Regulatory Life cycle
We help medical device companies in meeting the new, comprehensive requirements and compliance expectations during the entire lifecycle of their products all around the world.
.png)
Regulatory Lifecycle for Medical Devices
Medical devices are regulated around the world. But as medical device definitions varies in different parts of the world as well as classifications, activities required may vary. Therefore it is beneficial to plan your target markets in the regulatory plan(s) to be able to plan activities better. This allows smooth global registrations and reduced time to market. So if you are planning product for the US market and after that for the EU and Australia, it is worth to qualify and classify your product in all of these planned target markets. This is applicable especially if it looks like the device is not a medical device in your initial target market, for example in the US, but it might be somewhere else. It may affect heavily on the development activities. Labquality has experience of various markets.
Design
There are several common aspects in the technical documentations around the world, or could say, most aspects are the same. The most simplified pattern is:
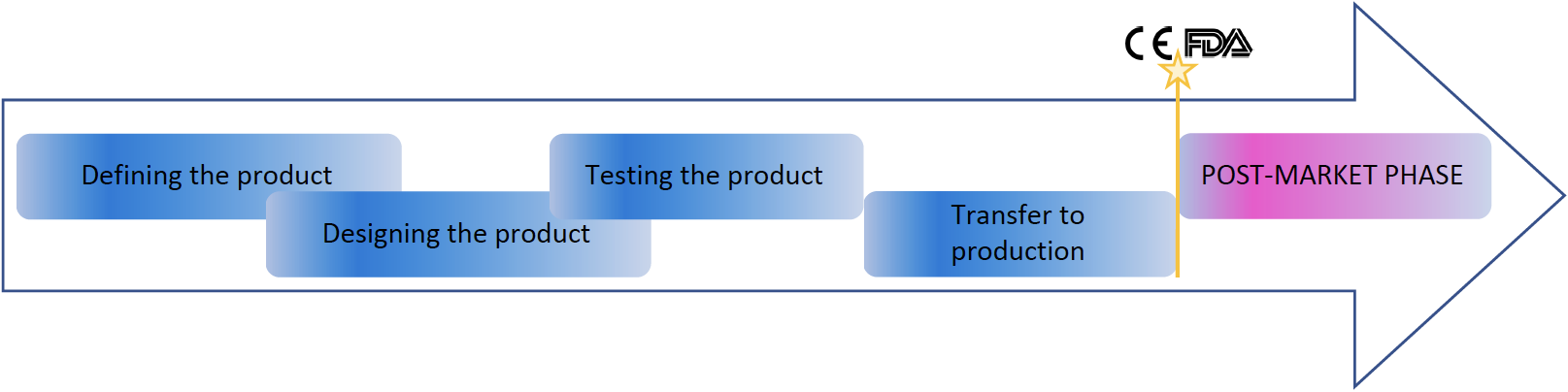
The whole design process must be covered with design and development (or design control) process of the manufacturing company. There requirements and risks are identified in the first place to understand what user wants, what are the requirements and related risks. This is similar everywhere. Although requirements and risks must include all the required aspects and the team thinking requirements and risks must have someone familiar with requirement and risk management processes, and Labquality have those experts to support you.
After defining phase, product is designed, tested and transferred to production. Each of these steps contain significant amount of documentation ie evidence, including specifications, test plans and reports, work instructions and device technical documentation. Significant part of the evidence for compliance is the clinical evidence. Especially EU regulations has increased such requirements, thus clinical evidence must be solid. This starts from clinical evaluation which often need to be supported with pre-market clinical investigation. Labquality can operate as your trusted CRO.
Regulatory lifecycle
After CE marking, FDA clearance or approval or other local sales approval, manufacturer must maintain technical documentation, inform changes to authorities where mandatory. Labquality has experts to monitor regulatory landscape changes and perform the activities needed to keep the medical devices on the desired markets. Most of the markets require active post market surveillance (depending on the risk level) and active reporting to authorities. Labquality provides also post market surveillance as a service (PMSaaS). So keeping product on multiple markets simultaneously require some work and organised mindset.
.jpg)
Labquality
How can we help?
Labquality can support you in
 planning regulatory pathways
planning regulatory pathways- documenting target market requirements to regulatory plan(s)
- performing qualification and classification for each target market
- supporting in design and development processes
- supporting in all technical documentation
- performing clinical evaluation planning and reporting
- clinical investigation planning, data management, execution and reporting
Explore our services

Electrical Safety IEC 60601
IEC 60601-1 is the basic safety standard for medical electrical devices and systems.
Clinical Investigation
Labquality provides services and expertise in the planning, preparation, and conduct of clinical investigations.
Biological Safety ISO 10993
We provide medical device manufacturers help with setting up a biological safety evaluation process and preparing biological evaluation documentation.
Qualification and classification of medical devices
Our team’s versatile experience and the available tools help in bringing clarity to the challenging cases.
Regulatory plan
Labquality can help medical device manufacturers in creating a regulatory plan.